Overview
![]()
Hammerhead is a tool designed to de novo identify bacterial DNA
methylation, including 4mC, 5mC, and 6mA, based on the strand-specific
mismatch patterns observed in R10.4.1 nanopore sequencing reads. It does
not require negative controls (methylation-free datasets); only native
DNA reads and references are needed as input for Hammerhead.
Workflow
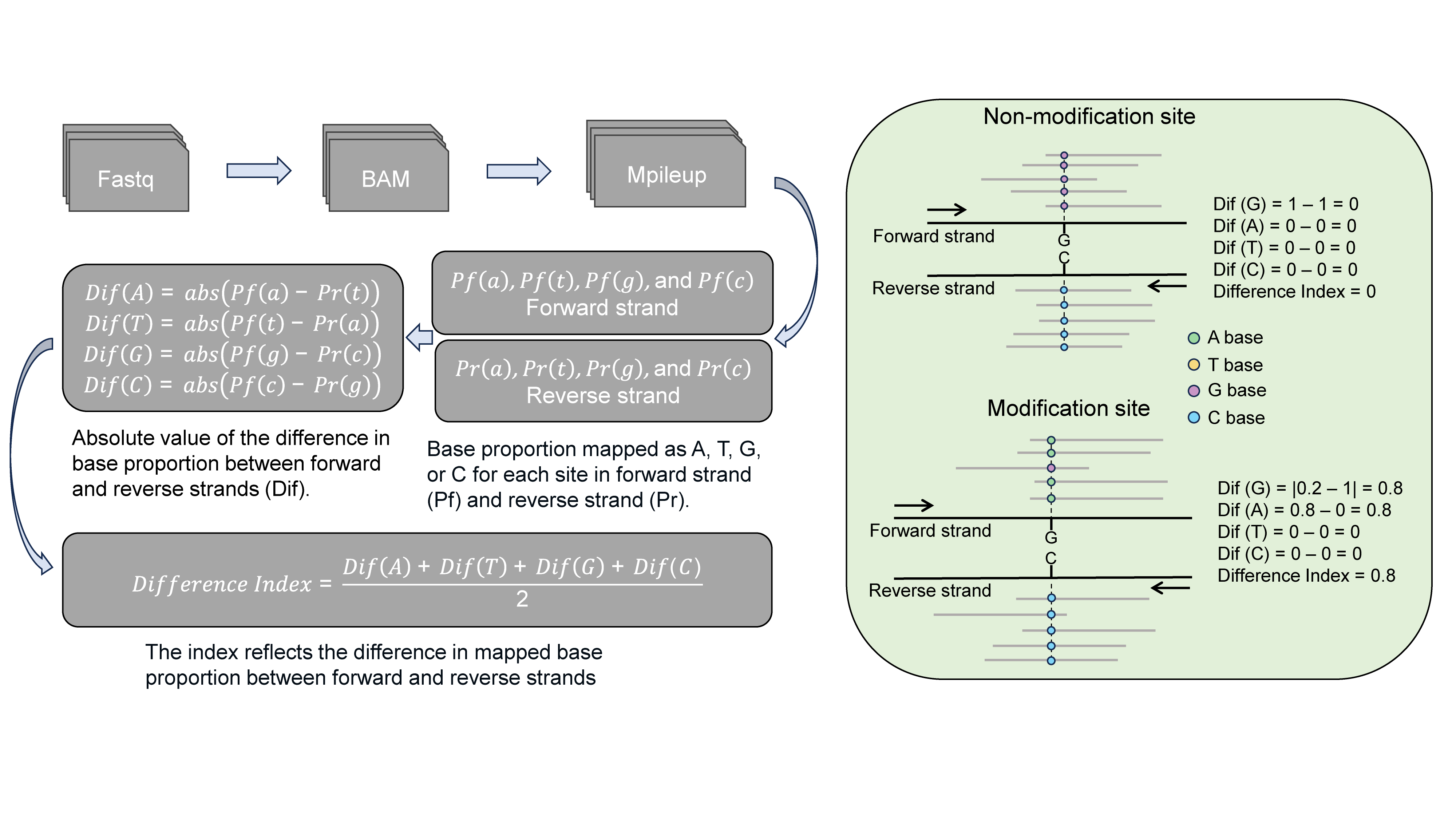
Hammerhead utilizes a self-defined metric called the difference
index (DI) to quantify the discrepancy in observed accuracy between the
forward and reverse strands at individual sites. This difference index
serves as a measure of the potential modification probability. A higher
value of the difference index indicates a higher likelihood of
modification at the corresponding site.
Installation
To use this tool, you’ll need to install additional tools or packages for read processing, including samtools and minimap2. The following command can help you install dependencies.
You can directly use Conda to download and install the dependencies based on the current environment.
conda install -c bioconda -c conda-forge minimap2 samtools bedtools -y
Also, you can create a new environment to install these dependencies.
conda create -n hammerhead -c bioconda -c conda-forge python samtools minimap2 bedtools -y
conda activate hammerhead
If you ncounter any version conflicts, you can select a specific version of the software to resolve the issue.
conda install -c bioconda -c conda-forge minimap2==2.17 samtools==1.17 bedtools==2.30.0 -y
Once you solve the problem of dependencies, the following command can
help to install the Hammerhead.
pip install Hammerhead-View
Quick usage
Hammerhead can be run in two different strategies to locate
methylation positions:
The first strategy is to select the sites with a difference index (DI) over the cutoff, the default is 0.35.
hammerhead --ref genome.fa --read input.fastq
The second strategy is to select the top N sites, based on the difference index sorted from the largest to the smallest, the default number is 2000.
hammerhead --ref genome.fa --read input.fastq --method top
Example
Here, we provide demo datasets (R10.4.1 simplex reads of E. coli) for
testing the Hammerhead. The following commands can help to download
them.
wget https://figshare.com/ndownloader/files/46437190 -O ecoli.fa
wget https://figshare.com/ndownloader/files/46437193 -O test.fastq.gz
Please run the following command to start data analysis!
hammerhead --ref ecoli.fa --read test.fastq.gz --min_depth 5 --min_depth_strand 3
Note: The arguments used in this command were for demonstration purposes only (the read coverage of data was too shallow) and may not reflect the optimal settings for your dataset. It is generally recommended to use the default arguments when you have sufficient read coverage, typically considered to be more than 50-fold coverage.
Detailed usage
usage: test [-h] --ref --read [--cpu] [--method] [--cut] [--num] [--min_depth] [--min_depth_strand]
A tool helps to find the potential modification sites
optional arguments:
-h, --help show this help message and exit
--ref Input reference (FASTA)
--read Input reads (FASTQ)
--cpu CPU number (default:10)
--method The strategy for detecting potential modification sites, cutoff or top (default:cutoff)
--cut Cutoff value [0, 1] (default:0.35)
--num Top N sites with the Difference index, sorted from the largest to the smallest (default:2000)
--min_depth The minimum depth (default:50)
--min_depth_strand The minimum depth for forward strand and reverse strand (default:25)
ref: the bacterial reference (FASTA format)
read: the R10.4.1 reads of the bacterial sample (FASTQ format), also support for the compressed data (sample.fastq.gz).
cpu: the number of CPUs used during analysis, the default is 10.
method: the methods to detect the potential modifications (cutoff or top), the default is the cutoff.
cut: the cutoff of DI, required by the cutoff in method, the default is 0.35.
num: the number of top sites, required by the top in method, the default is 2000.
min_depth: the minimum depth of the site used for downstream analysis, the default is 50.
min_depth_strand: the minimum depth of the forward or reverse site used for downstream analysis, the default is 25.
Strategy I: cutoff
When the cutoff is selected in the method argument, Hammerhead will
select the site with a DI over the cutoff (default: 0.35).
hammerhead --ref genome.fa --read input.fastq
The default value of 0.35 was selected based on the DI distribution of four methylation-free datasets to ensure a low false positive rate (FDR) of less than 1e-6. For some bacterial species with a different K-mer pattern with these four bacteria, we encourage the user to experiment with different cutoffs.
# examples
hammerhead --ref genome.fa --read input.fastq --cut 0.1
hammerhead --ref genome.fa --read input.fastq --cut 0.2
hammerhead --ref genome.fa --read input.fastq --cut 0.3
Strategy II: top
To avoid the cutoff issues, the top can be used in the method argument.
Hammerhead will select the top N sites based on the DI, sorted form
the largest to the smallest. The default number is 2000.
hammerhead --ref genome.fa --read input.fastq --method top
The user is also encouraged to experiment with different numbers.
hammerhead --ref genome.fa --read input.fastq --method top --num 1000
hammerhead --ref genome.fa --read input.fastq --method top --num 3000
Results
when you run the example provided by Hammerhead, you can get the following outputs.
├── ecoli.fa
├── ecoli.fa.fai
├── enrichment.bed
├── enrichment.fa
├── mapping.mpileup.txt
├── mapping.sort.bam
├── potential_modification_site.bed
├── potential_modification_site.txt
└── test.fastq.gz
Methylation files
The potential_modification_site.bed and potential_modification_site.txt are files that include the position and detailed metrics of potential modification sites. If using the top strategy, there will be an extra file named difference_index_table.txt, including the DI information for the total used sites. The following table is a demo of potential_modification_site.txt.
#Chr Start End Difference_index Dif_A Dif_T Dif_G Dif_C A,T,G,C,a,t,g,c
ecoli1_tig1 39570 39571 0.667 0.0 0.667 0.0 0.667 0,0,0,3,0,2,0,1
ecoli1_tig1 50133 50134 0.5 0.0 0.0 0.5 0.5 0,0,2,2,0,0,5,0
The Chr, strat, and End are the position information of potential modification sites. The Difference_index means the difference index, a metric indicates the probability of methylation. The Dif_A, Dir_T, Dir_G, and Dif_C mean the strand difference in A, T, G, and C bases, respectively. The A,T,G,C,a,t,g,c means the number of reads mapped as the A/T/G/C in the forward strand and T/A/C/G in the reverse strand.
Motif enrichment files
The enrichment.bed and enrichment.fa files are position and sequences of 21-mer reads. These reads are selected around the potential modification sites used for the motif enrichment analysis (-10 bp, +10 bp). The enrichment.fa file can be used for Meme, a website tool for motif enrichment, with a motif wide from 4 to 20 in advance options. Our own motif enrichment tool specific to Hammerhead is developing.
Other temporary files
The mapping.mpileup.txt and mapping.sort.bam files are temporary files generated during the read processing. You can remove them by following the command.
rm -r mapping.mpileup.txt mapping.sort.bam
Tool showcase
To show the potential of Hammerhead to identify the modifications in the bacterium. Here, two datasets from E. coli were used to call methylation including whole-genome sequencing (WGS) and whole-genome amplification (WGA) R10.4.1 simplex reads. The dam and dcm genes were found in the genome of the used E. coli strain. These two genes are associated with the G6mATC and C5mCWGG methylation.
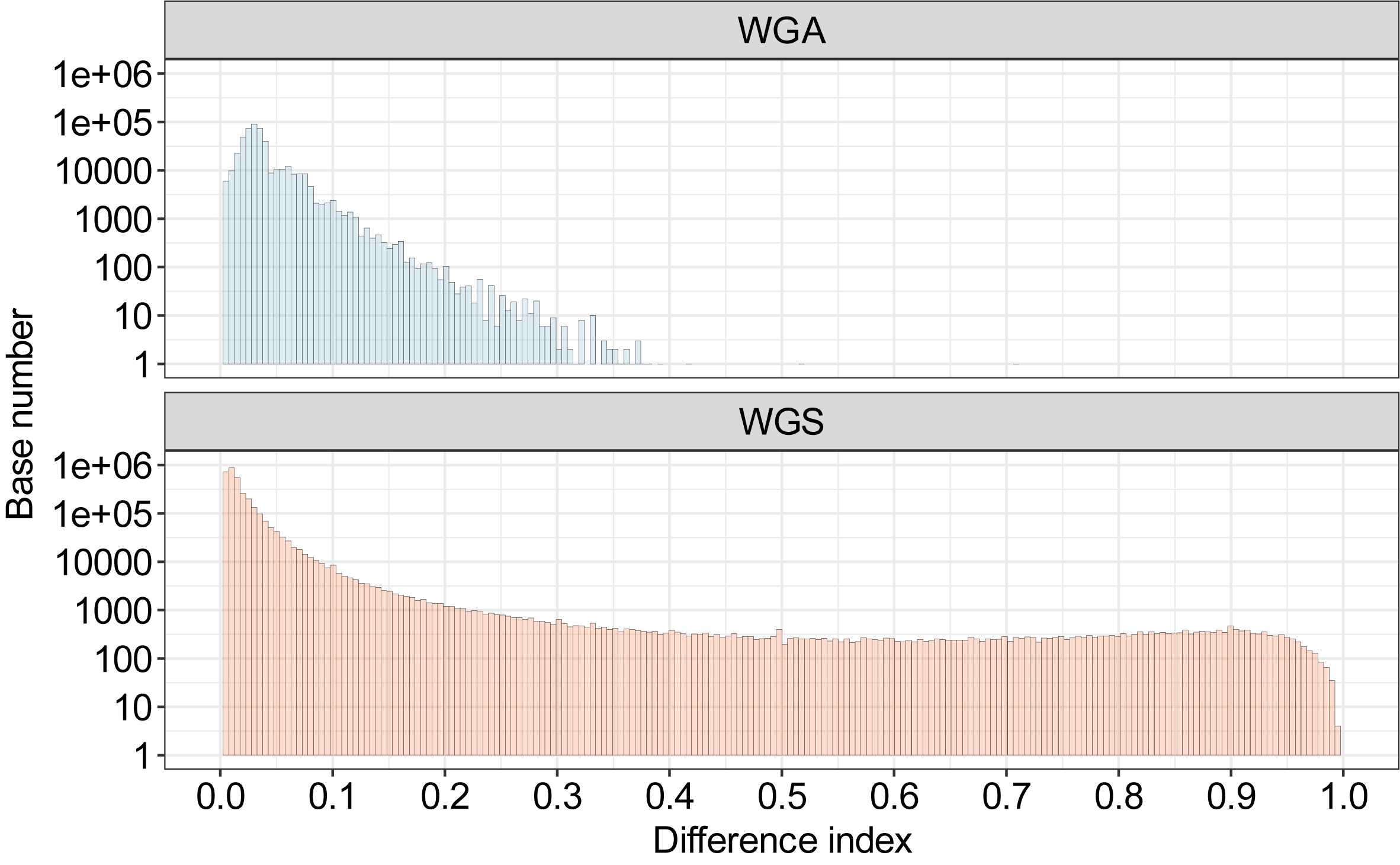
The distribution of difference index for sites in E. coli genome. The WGA reads were used as a negative control due to the lack of inherent methylation information. Based on the background noise of WGA reads, the sites with a difference index over 0.35 were regarded as potential modification sites.
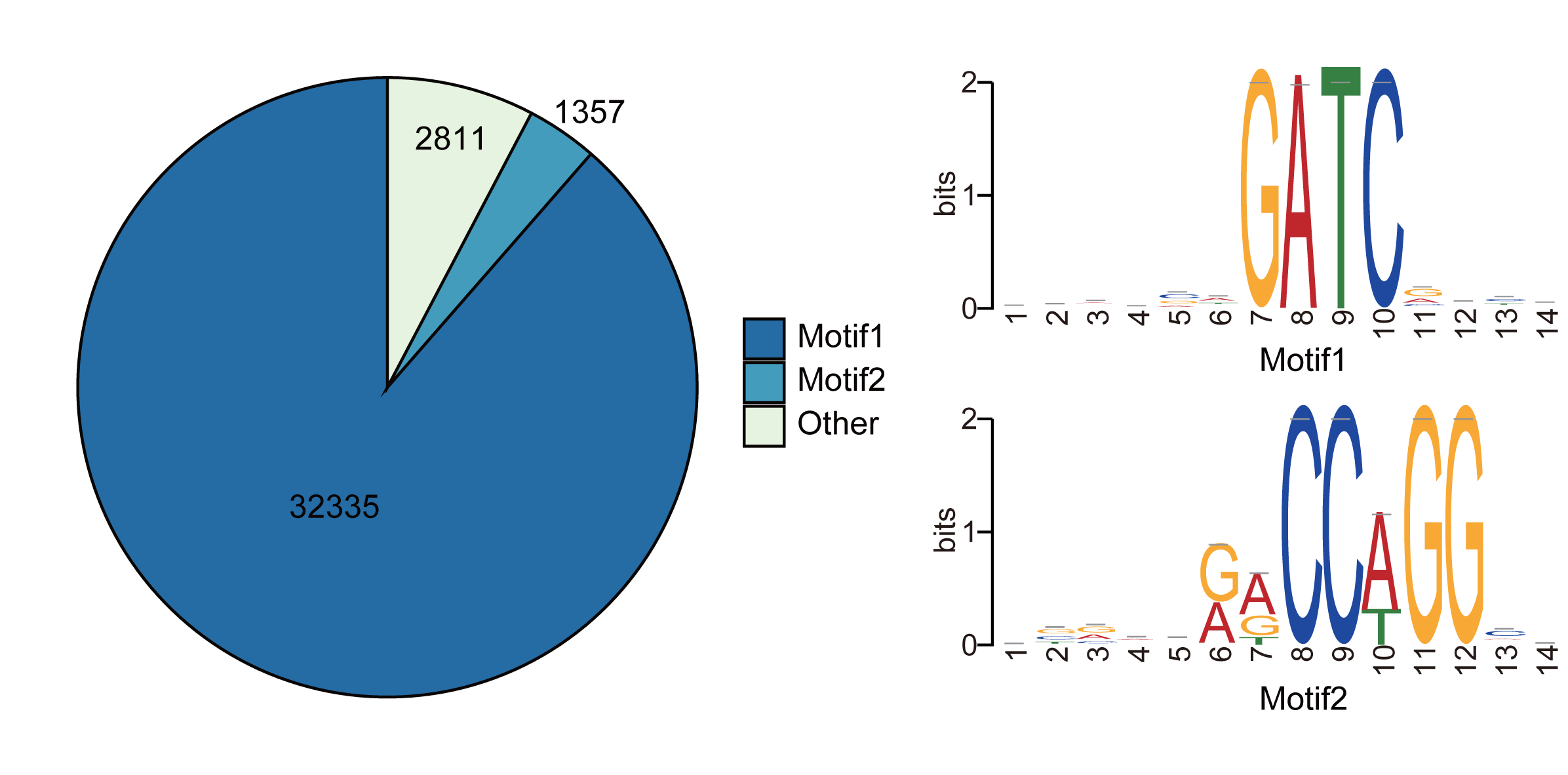
The motif of CCWGG and GATC was enriched using the sequences near these potential modification sites (-10 bp to +10 bp).
Note: Two datasets are available at the here. Both datasets were basecalled using the modification aware model, which is available in the part of the Retrained model.
Supplementary
Assemblies polish
Consider the presence of error-prone sites within ONT-based assemblies. Here we provided a pipeline to correct these sites by duplex polishing.
Step 1. Finding the potential modification sites
hammerhead --ref genome.fa --read sample.fq
After running the Hammerhead pipeline with your R10.4.1reads, you
will receive a file named potential_modification_site.bed. This file
includes the positions of the potential modification sites that have
been identified in your assembly.
Step 2. Polishing sites using duplex reads
hammer_polish --read <duplex_reads> --bed <position_sites> --ref <assembly>
After executing this script, you will obtain a file named
corrected_site.bed. This file contains information about the sites
that require replacement.
#the details of corrected_site.bed
#chr pos base N_A N_T N_G N_C P_A P_T P_G P_C polish_base
contig_4 29153 T 0 1 0 13 0.0 0.07142857142857142 0.0 0.9285714285714286 C
contig_6 155419 A 1 0 20 0 0.047619047619047616 0.0 0.9523809523809523 0.0 G
contig_7 60990 A 4 0 24 0 0.14285714285714285 0.0 0.8571428571428571 0.0 G *
contig_7 118348 G 25 0 7 0 0.78125 0.0 0.21875 0.0 A
contig_7 123180 C 0 19 0 2 0.0 0.9047619047619048 0.0 0.09523809523809523 T
contig_7 344011 C 0 19 0 4 0.0 0.8260869565217391 0.0 0.17391304347826086 T
chr, pos, base are the base type and position in the
reference of potential modification sites.
N_A, N_T, N_G, N_C are the number of reads which were
mapped as A, T, G, and C base at potential modification sites.
P_A, P_T, P_G, P_C are the proportion of reads which
were mapped as A, T, G, and C base at potential modification sites.
polish_base is the base after polished.
Note: The sites that are identified in the results are selected based on the proportion of errors. If you believe that any of the sites are correct, please manually remove them. The presence of an **asterisk (*)** indicates this is an ambiguous site that is challenging to determine as an error.
Step 3. Replacing erroneous bases with correct counterparts
cat corrected_site.bed | grep -v pos | awk '{if ($12 == "C") print $0}' | awk '{print $1 "\t" $2-1 "\t" $2}' > C.bed
cat corrected_site.bed | grep -v pos | awk '{if ($12 == "G") print $0}' | awk '{print $1 "\t" $2-1 "\t" $2}' > G.bed
cat corrected_site.bed | grep -v pos | awk '{if ($12 == "T") print $0}' | awk '{print $1 "\t" $2-1 "\t" $2}' > T.bed
cat corrected_site.bed | grep -v pos | awk '{if ($12 == "A") print $0}' | awk '{print $1 "\t" $2-1 "\t" $2}' > A.bed
bedtools maskfasta -fi <assembly> -bed A.bed -mc A -fo tmp1.fasta
bedtools maskfasta -fi tmp1.fasta -bed T.bed -mc T -fo tmp2.fasta
bedtools maskfasta -fi tmp2.fasta -bed G.bed -mc G -fo tmp3.fasta
bedtools maskfasta -fi tmp3.fasta -bed C.bed -mc C -fo final.fasta
# remove the temporary files
rm tmp1.fasta tmp2.fasta tmp3.fasta A.bed T.bed G.bed C.bed
The final.fasta is the final polished genome assembler.
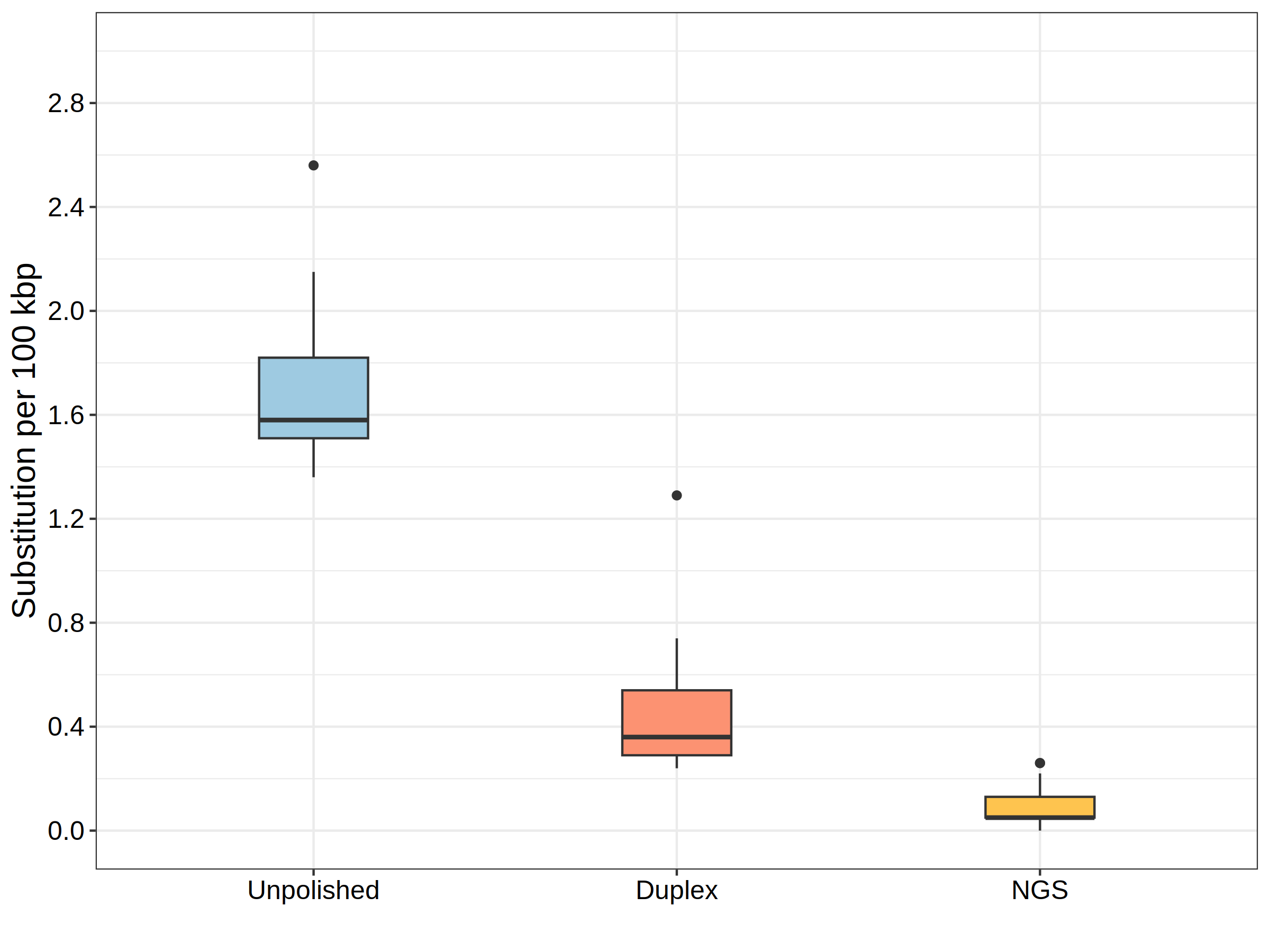
Tool showcase
To demonstrate the effectiveness of the polishing strategy based on the
Hammerhead in correcting substitution error types (G2A and C2T)
caused by DNA modifications in assemblies, we present the substitution
rates of 15 assemblies. These assemblies were generated using 40-, 50-,
and 60-fold random subsampling Acinetobacter pittii R10.4.1 reads. We
compared the results obtained from different polishing approaches with
the reference chromosome.
No polishing
Polishing potential modification sites with approximate 10-fold duplex reads
Polishing total assemblies with 50-fold next-generation sequencing (NGS) reads
Retrained model
Here we provide two retained models for users to help Hammerhead get
a higher sensitivity to modification like C5mCWGG. The new basecalling
model was fine-tuned from the original DNA super accuracy model (SUP)
using bonito (V0.7.2) with
the following parameters “–epochs 40 –lr 5e-4 –batch 32 –pretrained
dna_r10.4.1_e8.2_400bps_sup@v4.2.0”.
The model retrained with E. coli methylation-free data is available here.
The model retrained with four methylation-free data, including A. pittii, E. coli, E. faecium, and K. pneumoniae, is available here.